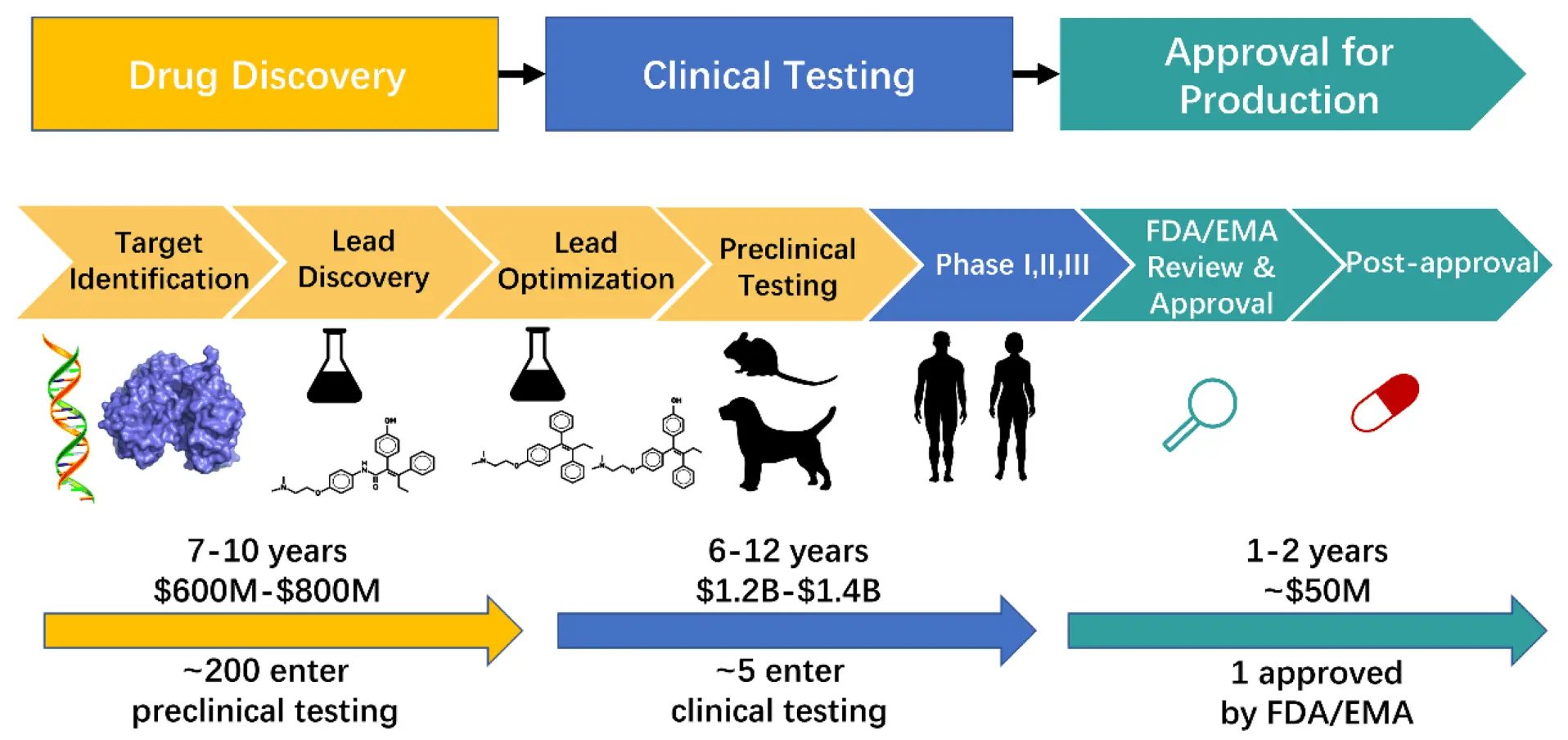
Drug Discovery
Main Approaches:
- Computational Chemistry using Approximations (HPC)
- AI based Methods
- Quantum Computing based Methods
The first two methods are unreliable quick and dirty approximations.
Only Quantum Computing can deliver accurate/perfect ab-initio solutions.
But there is a difference between Drug Discovery & Drug Design. Everyone in the world outside Automatski is doing the former. Automatski is the only one which can do the latter.
Drug Discovery - Techniques which can filter and select raw molecules from a database, that will bind to a specific target
Drug Design - On the other hand is ab-initio creation of molecules that can bind to a specific target, which can then be manufactured.